| Contents | Previous | Next |
Recent psychiatric literature has suggested that panic disorder is associated with a biophysiologic abnormality, as demonstrated by the observed familial predisposition (Crowe 1984), the positive treatment response to specific pharmacologic agents (Sheehan 1982), and the specificity of response to provocative tests (lactate, C02, caffeine, clonidine, yohimbine, hyperventilation) (Hollander et al. 1988). This chapter reviews a cognitive model of anxiety, the neuroanatomic structures hypothesized to be involved in the genesis of anxiety and fear, and the information derived from studies of patients with panic disorder during their basal state and during a variety of provocative tests.
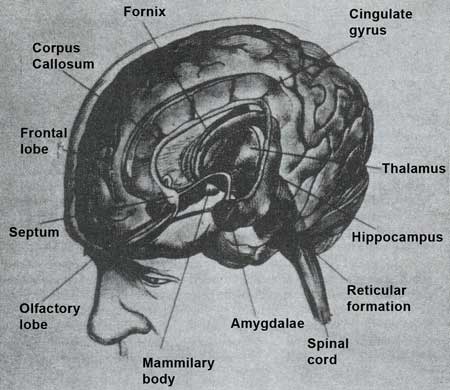
Although the brain structures and physiology involved in the sensations of anxiety and fear have not been entirely elucidated, a wealth of studies have greatly expanded current scientific knowledge. Both normal fear and abnormal anxiety responses involve specific deep brain structures more than the cerebral cortex (Marks 1987), specifically (1) the limbic system (hypothalamus, septum, hippocampus, amygdala, and cingulum); (2) other neuronal bodies including the thalamus, locus ceruleus, median raphe nuclei, and dental/interposital nuclei of the cerebellum; and (3) connections between these structures (figure 4).
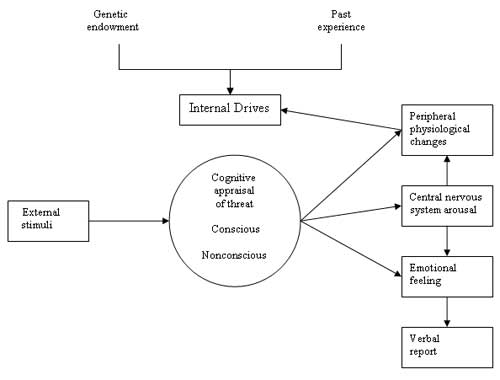
Figure 5 depicts a model of anxiety derived from the work of Sarason (1982), Spielberger et al. (1971), and Schacter and Singer (1962). In this model, both genetic endowment and emotional experiences in childhood determine the degree to which a person scans the environment for threat and cognitively appraises stimuli for potential danger (Lader 1983). Much of this scanning and appraisal is unconscious, and the cognitive process and resultant anxiety may seem puzzling to the individual when the reasons for the behavior have been forgotten. When a threat is perceived, an arousal process occurs, which in turn heightens vigilance and prepares the body for appropriate action (the fight or flight response). Fear is experienced with similar concomitant physiologic changes, which are considered adaptive and biologically advantageous when the precipitating stimulus is actually threatening. When the stimulus is not threatening or not apparent, however, the response is maladaptive.
| Figure 4. Specific deep brain structures involved in fear and anxiety |
|
|
| Source: Bloom etal. Brain. Mind, and Behavior. 2d ed. Copyright 1985, 1988 by the Educational Broadcasting System. Reproduced with permission of W.H. Freeman and Co. |
Beck (1976) stated that pathologic anxiety arises from several errors in cognitive appraisal, including: overestimating the probability of a feared event; overestimating the severity of a feared event; underestimating coping resources (what can be done about it); and underestimating reserve factors (what others can do to help). Cognitive studies of patients with panic disorder have found that these patients display a marked tendency to overestimate and exaggerate dramatic consequences to their somatic symptoms of anxiety. Thus, rapid heartbeat induced by exercise, or sweating induced by a crowded, warm room may lead to thoughts of loss of control or even death. These thoughts provoke more anxiety, arousal, and somatic symptoms, and a vicious cycle ensues (Rapee and Barlow 1988). Recently, the cognitive theory of panic has been used to explain nocturnal panic attacks whose occurrence had previously been used as evidence that panic disorder was a primarily biologic phenomenon (Roy-Byrne and Uhde 1988). Cognitive theorists have postulated that fluctuations in the internal physiologic state that occur during sleep may be sufficient to trigger anxiety and panic (Craske et al. 1989). Sleep of anxious patients has been characterized by a higher arousal level as shown by number of awakenings and changes in sleep stages. This cognitive model can be compared and contrasted to two prominent neurobiologic theories of anxiety: the septohippocampal theory and the locus ceruleus theory.
| Figure 5. A general model of anxiety |
|
|
| Source: Adapted from Lader, M. Behavior and anxiety: Physiologic mechanisms. Journal of Clinical Psychiatry 44(11-Sec. 2):7, 1983. Copyright 1983 by Physicians Post-Graduate Press, Inc. Reproduced with permission |
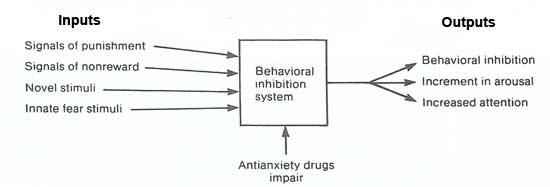
Gray (1982) proposed a complex theory of the neurobiology of anxiety that implicates the hippocampus and its afferent and efferent connections. According to his hypothesis, the septohippocampal system is primarily responsible for integrating and responding to novel or unpleasant environmental cues (figure 6).
| Figure 6. Model of the behavioral inhibition system |
|
|
| Source: Adapted from Lader, M. Behavior and anxiety: Physiologic mechanisms. Journal of Journal of Clinical Psychiatry 44(11-Sec. 2):8, 1983. Copyright 1983 by Physicians Post-Graduate Press, Inc. Reproduced with permission. |
The major inputs, and hence those that produce anxiety, are signals that warn of punishment or nonreward, innate fear stimuli, and novel stimuli. The major outputs are inhibition of motor behavior, increased level of arousal, and increased attention to the environment, especially to novel elements.
Gray suggested that the septohippocampal system itself predicts the next sensory event to which the organism will be exposed, checks whether it actually does occur, and then inhibits behavior if there is a mismatch, or if the predicted event is aversive. Although the septohippocampal system does not store information, it has access to and may modify information from the temporal lobe.
Gray also attempted to neuroanatomically localize the inputs and outputs of the system. He postulated that the ascending norepinephrine projection to the septohippocampal system determines whether incoming stimuli are important; the ascending norepinephrine system to the hypothalamus primes the effector autonomic, and motor systems for rapid action, if required; the ascending 5-HT projection adds information on whether this stimulus is associated with punishment; and the ascending cholinergic projection facilitates stimulus analysis. The prefrontal cortex and cingulate gyrus transmit to the septohippocampal system information for predicting sensory events and allow neocortical control of the septohippocampal system using verbally coded information.
Knowledge of this complex system is incomplete, but Gray's hypotheses have stimulated anxiety researchers to intensively study the involved neuroan-atomical systems. The septohippocampal model clearly has many similarities to the cognitive model presented earlier, with the septohippocampal system acting as an ongoing appraiser of threat to the organism (Lader 1983).
Supportive research has demonstrated that electrical stimulation of the hippocampus, parahippocampal gyrus, and amygdala most commonly produce sensations of fear in awake human subjects (Gloor et al. 1982). Moreover, PET research has shown that patients who are vulnerable to lactate-induced panic attacks have abnormally low ratios of left-to-right parahippocampal blood flow, blood volume, and oxygen metabolism in the resting nonpanic state (Reiman et al. 1986). Other researchers have found increased temporal lobe blood flow during panic in patients with panic disorder, as well as in controls with anticipatory anxiety (Curtis, unpublished paper). Research with the new MRI brain imaging technique (Fontaine et al. 1988) and brain electrical activity mapping (BEAM, a computerized spatial and temporal extension of conventional electroencephalography) (Abraham 1988) have also implicated temporal lobe abnormalities in panic disorder.
Numerous studies have demonstrated that the sympathetic nervous system is critically important in responding to stimuli that threaten the well-being of an organism (Cannon 1953; Heninger and Charney 1988). Activation of the sympathetic nervous system increases both norepinephrine and epinephrine. This section reviews studies describing the central control mechanism (locus ceruleus) of the sympathetic nervous system as well as those measuring peripheral catecholamines in patients with panic disorder.
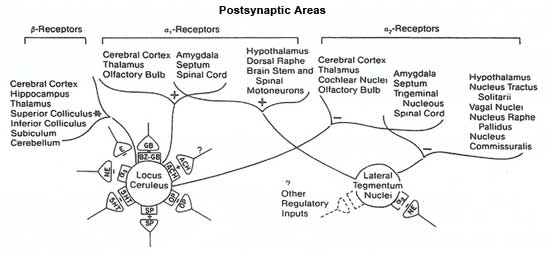
The hippocampal formation receives abundant noradrenergic projections from the locus ceruleus (Price and Marall 1981; Charney et al. 1984). Figure 7 illustrates the presynaptic regulation of the locus ceruleus and lateral tegmental nuclei and their axonal projections innervating postsynaptic alpha-1, alpha-2, and B-adrenergic receptors (Charney et al. 1984).
The locus ceruleus (LC) is a tiny blue (cerulear) streak in the dorsolateral tegmentum of the pons, comprising only about 400 neurons. The locus ceruleus contains nearly half the noradrenergic neurons and produces more than 70 percent of the total noradrenaline found in the brain (Marks 1987). Activation of the locus ceruleus has been associated with fear and alarm reactions in monkeys (Redmond 1976). Bilateral lesions of the locus ceruleus in animals were associated with failure to show normal cardioaccelerator responses to threatening stimuli and lack of the associated behavioral response (Snyder et al. 1977).
The locus ceruleus has projections to many regions of the brain associated with responses of pain and fear, and also to the cerebral cortex, which is probably involved with the interpretation of the importance or relevance of stimuli as well as their cognitive associations (Svensson 1987). The locus ceruleus also projects to limbic areas, such as the amygdala, which are important for emotional and cardiovascular control.
Reiman and colleagues (1986) postulated, based on PET scan data, that a parahippocampal abnormality determines vulnerability to anxiety attacks. They further hypothesized that a triggering event, such as activation of the locus ceruleus projections to the hippocampal formation, causes the abnormal region to initiate an anxiety attack by way of the septoamygdalar complex and its sequential projections.
| Figure 7. Brain noradrenergic function |
|
|
Schematized representation of complex neuroanatomy and neurophysiologic regulation of brain noradrenergic system. Presynaptic regulation of lucus ceruleus by a2 – adrenergic autoreceptors, and receptors for endogenous opiates, y-aminobutyric acid, substance P, acetylcholine, serotonin, and epinephrine is illustrated. Specific projection patterns of locus ceruleus and lateral tegmentum nuclei are shown in relation to type of adrenergic postsynaptic receptor innervated. Brain areas listed are those containing moderate to high levels of receptor specified. a2 indicates a2 -adrenergic autoreceptor; NE, norepinephrine; 5HT, serotonin; E, epinephrine; OP, opiate; ACH, acetylcholine; BZ-GB, benzodiazepine— γ-aminobutyric acid receptor complex; minus sign, inhibitory effect; plus sign, excitatory effect; asterisk, blocks frequency accommodation and enhances efficiency of sensory inputs. Source: Charney et al. Noradrenergic function in panic anxiety. Archives of General Psychiatry 41(8):760, 1984. Copyright 1984 by the American Medical Association. Reproduced with permission. |
The locus ceruleus not only monitors external, novel stimuli and controls defensive behavioral and autonomic responses to these stimuli, but also monitors internal stimuli from organs innervated by the autonomic nervous system (Svensson 1987). Thus, the LC monitors the peripheral cardiovascular and respiratory state (Elam et al. 1984) as well as the function of the digestive and urogenital systems (Elam et al. 1986). This biologic integrative system alerts the individual to stimuli important to survival, whether the stimulus is a threatening external situation (e.g., getting robbed at gunpoint) or an internal visceral or cardiovascular stimulus (e.g., a gastrointestinal hemorrhage or hypotension).
The activity of the locus ceruleus is regulated by neuronal systems, including the alpha-2 adrenergic autoreceptor, benzodiazepine receptors, endogenous opiates, serotonin, norepinephrine, epinephrine, acetylcholine, gammaaminobutyric acid (GABA), and substance P (Charney et al. 1984). Charney and colleagues (1984) have studied the alpha-2 adrenergic receptor, which is a major regulator of noradrenergic activity. A major function of the brain noradrenergic system may be to enhance the effects of sensory information to specific brain sites (Charney and Heninger 1986). The alpha-2 adrenergic receptor is located presynaptically on neuronal cell bodies or terminals, and it regulates the release of norepinephrine through a negative feedback mechanism (Cedarbaum and Aghajanian 1977; Andrade and Aghajanian 1984).
Other neurotransmitters also have important effects on noradrenergic neural activity. Epinephrine, serotonin, opiates, GABA, and glycine decrease the locus ceruleus’ rate of firing (Cedarbaum and Aghajanian 1977; Adrade and Aghajanian 1984; Segal 1979), while substance P, acetylcholine, and glutamate increase it (Guyenot and Aghajanian 1979). Drugs with anxiolytic effects such as benzodiazepines (Grant et al. 1983), tricyclic antidepressants (Svensson and Usdin 1978), morphine (Bird and Kuhar 1977), and the alpha-2 adrenergic agonist clonidine (Aghajanian 1978) depress locus ceruleus function, while drugs with anxiogenic effects, such as the alpha-2 adrenergic autoreceptor antagonist yohimbine, increase it (Holmberg and Gershon 1961; Charney et al. 1982).
The septohippocampal and locus ceruleus theories may be complementary or incompatible (Lader 1983). If the locus ceruleus functions to appraise external and internal stimuli, then it maybe equivalent to the septohippocampal system in Gray's model.
Charney and colleagues (1984, 1986) studied the central control of the autonomic nervous system by carefully examining the status of the alpha-2 adrenergic system in patients with panic disorder and controls. They gave provocative challenges of drugs that are agonists, such as clonidine (since the alpha-2 adrenergic receptor is inhibitory, an agonist should cause decreased LC activity), as well as antagonists such as yohimbine (which should cause increased LC activity). Clonidine produced significantly greater decreases in plasma 3-methoxy-4-hydroxyphenylglycol (MHPG — a metabolite of norepinephrine) levels and sitting and standing blood pressure, and significantly less self-rated sedation in patients with panic disorder than in controls (Charney et al. 1986). Yohimbine caused significantly increased MHPG levels, blood pressure, and behavioral responses of anxiety in patients with panic disorder when compared with controls (Charney et al. 1984). This increased oscillatory range of noradrenergic activity, observed as an increased sensitivity to both clonidine (agonist) and yohimbine (antagonist), may indicate that patients with panic disorder have an abnormally regulated alpha-2 noradrenergic receptor system (Roy-Byrne and Cowley 1988).
In addition to central control of the autonomic nervous system, peripheral catecholamines have been extensively studied in research on anxiety and fear. Patients with panic disorder were found to have increased heart rate, skin conductance, electromyelogram activity, and skin temperature associated with most anxiety attacks (Freedman et al. 1985; Taylor et al. 1985; Roth et al. 1985;
Shear 1986). Other researchers also found small increases in blood pressure, noradrenaline, plasma cortisol, and growth hormone during some attacks, but significant plasma epinephrine and MHPG changes were not observed (Cameron et al. 1987). Charney and colleagues (1984) found that MHPG was higher in patients with panic disorder who had frequent panic attacks than in normal controls or depressives.
At rest, panic disorder patients have tachycardia and heart rates that are more variable than controls (Taylor et al. 1985). Several investigators found increased plasma levels of epinephrine in the resting or basal state in patients with panic disorder (Villacres et al. 1987; Nesse et al. 1984; Appleby et al. 1981; Ballenger et al. 1984). Although early studies suggested plasma norepinephrine might also be elevated in anxious patients (Wyatt et al. 1971; Mathew et al. 1980; Ballenger et al. 1984), later studies found no such elevations (Villacres et al. 1987; Appleby et al. 1981; Carr et al. 1984; Nesse et al. 1984; Liebowitz et al. 1985).
Although the racing and pounding heartbeats, shortness of breath, sweating, rapid and deep breathing, and chest pain that typically occur during a panic attack suggest beta-adrenergic activation, as does the fact that tricyclic antidepressants, which are effective in treating panic disorder, down-regulate or decrease central beta-adrenergic receptor sensitivity (Charney et al. 1981a, b), recent evidence suggests that beta-adrenergic function may be decreased rather than increased in panic disorder (Nesse et al. 1984). Two studies of responses to infusions of isoproterenol (a beta-adrenergic agonist) have been carried out in patients with panic disorder (Nesse et al. 1984; Rainey et al. 1984). In one study, isoproterenol caused more panic attacks in patients with panic disorder than in controls, although the anxiety produced was less intense than with lactate infusion and no tachycardia occurred (Rainey et al. 1984a, b). In the second study, no increase in panic attacks was found in panic disorder patients, although the dosage of isoproterenol used was one-third of that in the first study (Nesse et al. 1984). The latter study actually showed decreased physiologic responsiveness to isoproterenol (decreased heart rates) in patients with panic disorder compared to controls. These two studies do not strongly support a betaadrenergic abnormality in panic disorder. They are consistent with well controlled studies showing that beta-blockers have only modest antianxiety effects and are not specifically effective in blocking panic attacks. For example, intravenously administered propranolol, in dosages sufficient to cause full beta-adrenergic blockade, is not able to block lactateinduced panic attacks in patients with panic disorder (German et al. 1983).
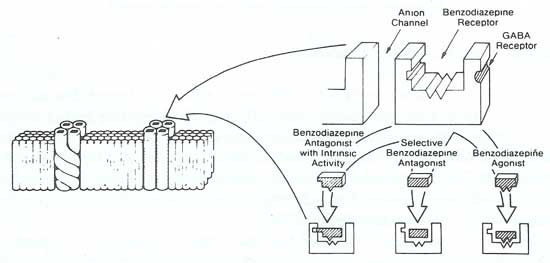
An exciting finding in anxiety research was the discovery of brain benzodiazepine (Bz) receptors (Insel et al. 1984). The Bz receptors are linked to a receptor for the inhibitory neurotransmitter GABA (figure 8) (Skolnick and Paul 1983). Binding of a benzodiazepine to the Bz receptor facilitates the action of GABA, which is known to increase the permeability of chloride ions through the chloride ion channel. The heightened permeability of the cell to chloride ions decreases neuronal excitability by hyperpolarizing the neuronal membrane (Paul 1988). These diffuse, inhibitory short GABA-ergic circuits and associated Bz receptors are present throughout the brain and spinal cord. GABA and benzodiazepine have inhibitory effects on the locus ceruleus (Grant et al. 1980) and may also act to decrease anxiety by modulating the ascending activating systems (serotonergic, noradrenergic, and probably dopaminergic) that are implicated in the expression of fear (Marks and Tobena 1986).
There also appear to be endogenous anxiogenic compounds (betacarbolines and purines) that act on Bz receptors. Thus, these receptors may mediate both increases and decreases in anxiety (Roy-Byrne and Cowley 1988). Roy-Byrne and Veith (unpublished observations) found that panic patients may be less sensitive to the catecholamine reducing effects of anxiolytic benzodiazepines. Woods, Charney, and Silver (1988a) reported that patients with panic disorder appear more sensitive to the effect of benzodiazepines with anxiogenic properties. Taken together, these results suggest that an abnormality of the Bz receptor may be present in patients with panic disorder.
Long-term alprazolam treatment of patients with panic disorder has been shown to reduce norepinephrine turnover, as evidenced by decreased baseline levels of the norepinephrine metabolite MHPG in plasma and the blunted yohimbine-induced increases in plasma MHPG, anxiety, and blood pressure (Charney and Heninger 1985). Other studies, however, suggested that alprazolam decreases the firing rate of the locus ceruleus only a small amount compared with imipramine or desipramine (Charney et al. 1981a). Gray (1977) postulated that rather than having their primary effect on noradrenergic systems, benzodiazepines enhance recurrent inhibition within the hippocampus (dampen input) or enhance feedback inhibition of both the septal and hippocampal systems.
| Figure 8. Model for GABA-receptor, benzodiazepine binding site, anion channel complex |
|
|
| Source: Insel et al. Benzodiazepine receptor-mediated model of anxiety. Archives of General Psychiatry 41 (8):747, 1984. Copyright 1984 by the American Medical Association. Reproduced with permission. |
Exhibit 10 lists the provocative laboratory studies carried out in recent years comparing patients with panic disorder and controls. These provocative tests enabled researchers to study in the laboratory the biophysiologic changes that occur during a panic attack and provided a conceptual framework for understanding complex phenomena (Uhde and Tancer 1988). Although the provocative tests fail to replicate all components of the panic attack, they have given researchers important information about the brain structures involved in these anxiety attacks.
Exhibit 10. Provocative tests in patients with panic disorder |
Sodium lactate Hyperventilation |
Several researchers in the late 1940s and early 1950s found that patients with effort intolerance were distinguished from controls by their higher oxygen consumption with vigorous exercise and greater serum lactate production (Cohen and White 1950; Holmgren and Strom 1959; Jones and Mellerish 1946; Linko 1950). The increased serum lactate noted on exercise was hypothesized to be related to an increased muscle oxygen debt and a shift to an anaerobic metabolism (Ackerman and Sachar 1974).
This finding stimulated Pitts and McClure (1967) to speculate that patients with panic attacks were more sensitive to the effects of lactate than controls. They found that administration of 10 mg/kg of 0.5 molar sodium lactate infused over 20 minutes induced panic attacks in about 75 percent of patients with panic disorder and less than 5 percent of controls. This effect has now been replicated numerous times (Liebowitz et al. 1985; Cowley et al. 1987o; Rainey et al. 1984b; Margraf et al. 1986). Panic attacks are induced only in patients with panic disorder and not in patients with other psychiatric diagnoses (obsessive-compulsive disorder, major depression, social phobia, or bulimia) (Cowley et al. 1987b; Uhde and Tancer 1988). However, the interpretation of these findings in developing an overall theory of the genesis of panic attacks remains controversial (Margraf et al. 1986).
Several theories have been proposed to explain the association of sodium lactate infusion with panic attacks. The first suggests that sodium lactate produces nonspecific arousal and uncomfortable bodily sensations in all patients (both with and without panic disorder), but that the patients with panic disorder (owing to their hypervigilance) overreact to these sensations, as they do to many stimuli. Liebowitz and colleagues (1984) found that the higher the initial anxiety level, diastolic blood pressure, and heart rate of patients with panic disorder, the greater the likelihood of developing an anxiety attack with sodium lactate infusion. This suggests an interaction between the initial level of arousal, cognitive expectancy, and the effects of lactate (Margraf et al. 1986). Other evidence against lactate itself being a biochemical cause of panic is that moderate to strenuous exercise in normals frequently causes serum lactate to rise to levels similar to those resulting from lactate infusion without producing any anxiety (Ackerman and Sachar 1974).
Another theory suggests that the conversion of lactate to bicarbonate and C02 produces a transient intracerebral hypercapnia (causing stimulation of C02 brainstem chemoreceptors), since C02, but not bicarbonate, crosses the blood-brain barrier (Carr and Sheehan 1984). This theory is supported by the finding that patients with panic disorder are more sensitive to air with increased C02 concentrations than are controls (German et al. 1986c).
Finally, Rainey and colleagues (1985) hypothesized that panic disorder results from a subtle defect in aerobic metabolism. This hypothesized defect explains the increased level of lactate seen with exercise, the increase in oxygen consumption and lactate levels in patients with panic disorder versus controls who are infused with lactate, and the panic response to a variety of other challenges (yohimbine, caffeine, and CO2) that increase metabolic demand.
The two main physiologic differences consistently found in patients with lactate induced panic are hyperventilation and tachycardia (Liebowitz et al. 1985). Both metabolic and respiratory alkalosis develop in all subjects during lactate infusion, but only hyperventilation-induced hypocapnia differentiates patients at the point of lactate-induced panic from nonpanicking patients and normal controls (Liebowitz et al. 1985). Low inorganic phosphate levels at baseline also appear associated with patients who panic during lactate infusion (German et al. 1986a). Other than this, few consistent biochemical changes differentiate patients who panic during lactate infusion and controls who do not (Carr et al. 1986).
Researchers have demonstrated that when panic disorder patients are successfully treated with either imipramine or alprazolam and then retested with lactate infusion, most patients no longer develop panic attacks. This suggests a protective biologic change induced by medication in these patients (Roy-Byrne and Cowley 1988).
Studies of patients with soldier’s heart suggested that these patients displayed intolerable hyperpnea at lower levels of C02 than normals (Drury 1987). In the 1950s, patients with neurocirculatory asthenia (a syndrome that is also virtually indistinguishable from panic disorder) were found to have a greater increase in ventilation in response to muscular work than normals and were more likely to have anxiety attacks with 4-percent C02 than were controls (Cohen and White 1950). Patients with hyperventilation syndrome have also been reported to have abnormalities in the C02 ventilatory response to carbon dioxide inhalation (Folgering and Colla 1978).
Several studies have now demonstrated that a significantly higher number of patients with panic disorder than controls have anxiety attacks with 5-percent carbon dioxide inhalation (Gorman et al. 1984, 1986c; Woods et al. 1988b; Gorman et al. 1988a). Room air contains virtually no C02 (0.5 percent). Five-percent C02 in room air will triple minute ventilation in most normal subjects (Mitchell et al. 1963), and concentrations of C02 over 12 percent induce central nervous system and respiratory depression (Berger et al. 1977). Woods and colleagues (1988&) found that, although the frequency of panic attacks and the increases in anxiety and somatic symptoms induced by 5-percent C02 were greater in panic attack patients than in controls, 7.5-percent C02 induced quite similar severe anxiety and somatic symptoms in the healthy controls.
Panic with C02 has been associated with an exaggerated ventilatory response and increases in plasma norepinephrine and diastolic blood pressure (Gorman et al. 1988a). Gorman postulated that patients with panic disorder may have hypersensitive C02 receptors or C02 receptors with low setpoints. When triggered by increasing C02, these receptors evoke a subjective panic associated with an exaggerated ventilatory response and consequent hypocapnic alkalosis. Patients may "learn" to hyperventilate to maintain low levels of C02 and avoid triggering their C02 sensors; this would explain the previous finding that patients with panic disorder are chronic hyperventilators (Liebowitz et al. 1985).
Alternatively, increasing C02 may cause panic by stimulating the locus ceruleus (Woods et al. 1985, 1988b; Redmond 1979). Carbon dioxide increases brain catecholamine synthesis (Carlsson et al. 1977; Garcia de Yebenes Prous et al. 1977) and norepinephrine turnover (Garcia de Yebenes Prous et al. 1977) in the rat, increases the firing rate of neurons in the rat noradrenergic locus ceruleus (Elam et al. 1981), and increases the plasma levels of the norepinephrine metabolite MHPG in the rhesus monkey. Also, lesions in the locus ceruleus have reduced by 30 to 80 percent the C02-induced increase in brain biogenic amine release (Oke et al. 1983).
A final hypothesis states that, while breathing increased C02 causes increased somatic symptoms in all persons, the hypervigilance (increased alertness to subtle changes in their physiologic status) and tendency to cognitively catastrophize of patients with panic disorder leads to a conditioned hyperresponse (dark 1986a; Evans 1972). This is supported by a recent study of patients with panic disorder that demonstrated that having an illusion that they could control the amount of carbon dioxide they were administered led to a reduction in the number of DSM-III-R panic attack symptoms, a rating of less intense symptoms, a report of less subjective anxiety attacks and catastrophic cognitions, and significantly less anxiety when exposed to C02 compared to subjects without an illusion of control (Sanderson et al. 1989).
Caffeine is a xanthine derivative and is a widely used psychotropic agent in North America, with daily consumption averaging 200 to 250 mg per adult (Gilbert 1981). In short-term administration, caffeine increases alertness, stimulates attention, and helps restore performance that is decreased by factors such as boredom and fatigue (Boulenger et al. 1984; Weiss and Laties 1962). Caffeine can, in doses above 600 mg/day, induce a syndrome of caffeinism. It is characterized by nervousness, anxiety, sleep disturbance, and psychophysiologic symptoms (rapid heartbeat, chest tightness, dyspnea) that may be indistinguishable from panic disorder (Greden 1974).
Several researchers have found that patients with panic disorder consume less caffeine than normal control subjects or patients with major depression, presumably because of its anxiogenic effect (Boulenger et al. 1984; Lee et al. 1988). Greater sensitivity to caffeine among panic disorder patients has been shown by direct caffeine challenge (Charney et al. 1985; Uhde et al. 1985&). Charney and colleagues (1985) also demonstrated that caffeine (10mg/kg) produced significantly greater increases in subject-rated anxiety, nervousness, fear, nausea, palpitations, restlessness, and tremors in patients with panic disorder compared to healthy controls. These effects in panic disorder patients were significantly correlated with plasma caffeine levels, and almost three qarters of the panic disorder patients reported that the behavioral effects of caffeine were similar to those experienced during panic attacks. Caffeine did not alter MHPG levels in patients or controls and increased plasma cortisol equally in these two groups. Following caffeine challenge, panic patients had higher plasma lactate levels than did controls, suggesting that caffeine does have a metabolic stimulating effect and may have a mechanism of action similar to lactate.
Several mechanisms of action for caffeine have been proposed, including inhibition of phosphodiesterase (Butcher and Sutherland 1962), antagonism of benzodiazepine (Marangos et al. 1979) and adenosine receptor function (Snyder and Sklar 1984), and increased brain catecholamine activity (Berkowitz et al. 1970). The inhibition of adenosine receptor function is currently favored as the mechanism for caffeineinduced anxiety and stimulation (Snyder and Sklar 1984; Charney et al. 1985), because the brain concentration of caffeine required to block adenosine receptors is within the range seen following normal doses of caffeine.
There is considerable evidence that adenosine and other purines act as neuromodulators with important regulatory effects on multiple neuronal systems throughout the brain (Charney et al. 1985; Stone 1981; Patel et al. 1982). Adenosine potently inhibits the release of acetylcholine (Ginsborg and Hirst 1972) and norepinephrine (Enero and Saidman 1977) centrally, and caffeine antagonizes the effects of adenosine on neurotransmitter release and causes increased norepinephrine and acetylcholine release (Stone 1981). Adenosine has been shown by electrophysiologic studies to reduce the spontaneous firing rate of neurons in the cerebral cortex, cerebellum, and locus ceruleus (Oipe et al. 1983). These inhibitory effects of adenosine occur via interactions with adenosine receptors and are blocked by caffeine (Charney et al. 1985). Thus, caffeine and other methylanthines, such as theophylline and isobutylmethylxanthine, increase the firing of cortical neurons and the locus ceruleus and may produce their stimulatory effects by this mechanism.
The plethora of provocative tests of panic disorder, the research on brain receptors, and the recent MRI, PET scan, and BEAM studies have produced an exponential growth of knowledge on the psychobiology of panic disorder and other types of anxiety. However, the complexity of the brain has thus far prevented the full elucidation of brain mechanisms involved in anxiety. As in many other medical illnesses, treatment has preceded full understanding of pathophysiology. Also, analogous to medical illnesses such as peptic ulcer disease, knowledge of factors that provoke a relapse of symptoms (such as alcohol, spicy foods, or coffee in peptic ulcer disease) does not necessarily clarify the pathophysiologic mechanisms in that disease. Future biologic research promises not only to unravel the enigma of pathologic anxiety, but also to add to our knowledge of overall brain physiologic function.